PlotResults
AG Chiocchetti
29 Januar 2021
Last updated: 2021-09-24
Checks: 7 0
Knit directory: femNATCD_MethSeq/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20210128) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 56bb68f. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.Rhistory
Ignored: code/.Rhistory
Ignored: data/Epicounts.csv
Ignored: data/Epimeta.csv
Ignored: data/Epitpm.csv
Ignored: data/KangUnivers.txt
Ignored: data/Kang_DataPreprocessing.RData
Ignored: data/Kang_dataset_genesMod_version2.txt
Ignored: data/PatMeta.csv
Ignored: data/ProcessedData.RData
Ignored: data/RTrawdata/
Ignored: data/SNPCommonFilt.csv
Ignored: data/femNAT_PC20.txt
Ignored: output/BrainMod_Enrichemnt.pdf
Ignored: output/Brain_Module_Heatmap.pdf
Ignored: output/DMR_Results.csv
Ignored: output/GOres.xlsx
Ignored: output/LME_GOplot.pdf
Ignored: output/LME_Results.csv
Ignored: output/LME_Results_Sig.csv
Ignored: output/LME_tophit.svg
Ignored: output/ProcessedData.RData
Ignored: output/RNAvsMETplots.pdf
Ignored: output/Regions_GOplot.pdf
Ignored: output/ResultsgroupComp.txt
Ignored: output/SEM_summary_groupEpi_M15.txt
Ignored: output/SEM_summary_groupEpi_M2.txt
Ignored: output/SEM_summary_groupEpi_M_all.txt
Ignored: output/SEM_summary_groupEpi_TopHit.txt
Ignored: output/SEM_summary_groupEpi_all.txt
Ignored: output/SEMplot_Epi_M15.html
Ignored: output/SEMplot_Epi_M15.png
Ignored: output/SEMplot_Epi_M15_files/
Ignored: output/SEMplot_Epi_M2.html
Ignored: output/SEMplot_Epi_M2.png
Ignored: output/SEMplot_Epi_M2_files/
Ignored: output/SEMplot_Epi_M_all.html
Ignored: output/SEMplot_Epi_M_all.png
Ignored: output/SEMplot_Epi_M_all_files/
Ignored: output/SEMplot_Epi_TopHit.html
Ignored: output/SEMplot_Epi_TopHit.png
Ignored: output/SEMplot_Epi_TopHit_files/
Ignored: output/SEMplot_Epi_all.html
Ignored: output/SEMplot_Epi_all.png
Ignored: output/SEMplot_Epi_all_files/
Ignored: output/barplots.pdf
Ignored: output/circos_DMR_tags.svg
Ignored: output/circos_LME_tags.svg
Ignored: output/clinFact.RData
Ignored: output/dds_filt_analyzed.RData
Ignored: output/designh0.RData
Ignored: output/designh1.RData
Ignored: output/envFact.RData
Ignored: output/functional_Enrichemnt.pdf
Ignored: output/gostres.pdf
Ignored: output/modelFact.RData
Ignored: output/resdmr.RData
Ignored: output/resultsdmr_table.RData
Ignored: output/table1_filtered.Rmd
Ignored: output/table1_filtered.docx
Ignored: output/table1_unfiltered.Rmd
Ignored: output/table1_unfiltered.docx
Ignored: setup_built.R
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the repository in which changes were made to the R Markdown (analysis/04_Plot_Results.Rmd) and HTML (docs/04_Plot_Results.html) files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view the files as they were in that past version.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| html | 43333cc | achiocch | 2021-09-24 | adds new build |
| html | b497cc9 | achiocch | 2021-09-17 | Build site. |
| html | ccbc9e4 | achiocch | 2021-08-10 | Build site. |
| html | 70cd649 | achiocch | 2021-08-06 | Build site. |
| html | 2a53a87 | achiocch | 2021-08-06 | Build site. |
| Rmd | e4425b8 | achiocch | 2021-08-06 | wflow_publish(c(“analysis/", "code/”, "docs/*")) |
| html | e3a9ae3 | achiocch | 2021-08-04 | Build site. |
| html | f6bbdc0 | achiocch | 2021-08-04 | Build site. |
| html | 710904a | achiocch | 2021-08-03 | Build site. |
| html | 16112c3 | achiocch | 2021-08-03 | wflow_publish(c(“analysis/", "code/”, “docs/”)) |
| html | cde8384 | achiocch | 2021-08-03 | Build site. |
| Rmd | d3629d5 | achiocch | 2021-08-03 | wflow_publish(c(“analysis/", "code/”, "docs/*")) |
| html | d761be4 | achiocch | 2021-07-31 | Build site. |
| html | b452d2f | achiocch | 2021-07-30 | wflow_publish(c(“analysis/", "code/”, "docs/*")) |
| html | 2734c4e | achiocch | 2021-05-08 | Build site. |
| html | a847823 | achiocch | 2021-05-08 | wflow_publish(c(“analysis/", "code/”, "docs/*")) |
| html | 9cc52f7 | achiocch | 2021-05-08 | Build site. |
| html | 158d0b4 | achiocch | 2021-05-08 | wflow_publish(c(“analysis/", "code/”, "docs/*")) |
| html | 0f262d1 | achiocch | 2021-05-07 | Build site. |
| html | 5167b90 | achiocch | 2021-05-07 | wflow_publish(c(“analysis/", "code/”, "docs/*")) |
| html | 05aac7f | achiocch | 2021-04-23 | Build site. |
| html | f5c5265 | achiocch | 2021-04-19 | Build site. |
| Rmd | dc9e069 | achiocch | 2021-04-19 | wflow_publish(c(“analysis/", "code/”, "docs/*")) |
| html | 17f1eec | achiocch | 2021-04-10 | Build site. |
| html | 91de221 | achiocch | 2021-04-05 | Build site. |
| Rmd | b6c6b33 | achiocch | 2021-04-05 | updated GO function, and model def |
| html | 4ea1bba | achiocch | 2021-02-25 | Build site. |
| Rmd | 6c21638 | achiocch | 2021-02-25 | wflow_publish(c(“analysis/", "code/”, "docs/*"), update = F) |
| html | 6c21638 | achiocch | 2021-02-25 | wflow_publish(c(“analysis/", "code/”, "docs/*"), update = F) |
Home = getwd()Dark8 = brewer.pal(8, "Dark2")
source(paste0(Home,"/code/custom_functions.R"))Lade nötiges Paket: kableExtraLade nötiges Paket: tidyverse-- Attaching packages --------------------------------------- tidyverse 1.3.0 --v ggplot2 3.3.3 v purrr 0.3.4
v tibble 3.0.4 v dplyr 1.0.2
v tidyr 1.1.2 v stringr 1.4.0
v readr 1.4.0 v forcats 0.5.0-- Conflicts ------------------------------------------ tidyverse_conflicts() --
x dplyr::collapse() masks IRanges::collapse()
x dplyr::combine() masks Biobase::combine(), BiocGenerics::combine()
x dplyr::count() masks matrixStats::count()
x dplyr::desc() masks IRanges::desc()
x tidyr::expand() masks S4Vectors::expand()
x dplyr::filter() masks stats::filter()
x dplyr::first() masks S4Vectors::first()
x dplyr::group_rows() masks kableExtra::group_rows()
x dplyr::lag() masks stats::lag()
x ggplot2::Position() masks BiocGenerics::Position(), base::Position()
x purrr::reduce() masks GenomicRanges::reduce(), IRanges::reduce()
x dplyr::rename() masks S4Vectors::rename()
x dplyr::slice() masks IRanges::slice()Lade nötiges Paket: compareGroupsLade nötiges Paket: scales
Attache Paket: 'scales'The following object is masked from 'package:purrr':
discardThe following object is masked from 'package:readr':
col_factorload(paste0(Home,"/output/dds_filt_analyzed.RData"))
load(paste0(Home,"/output/resultsdmr_table.RData"))
Patdata=colData(dds_filt)
cpm=counts(dds_filt, normalized=T)
log2_cpm=log2(cpm+1)
res=results(dds_filt)Plot significant LME hits
GRresultslme_table = rowRanges(dds_filt)
DFtoplotall=as.data.frame(GRresultslme_table)
colnames(DFtoplotall)[1:3] = c("Chromosome", "chromStart", "chromEnd")
DFtoplotall = DFtoplotall[order(DFtoplotall$Chromosome, DFtoplotall$chromStart),]
DFtoplotall$Chromosome = as.factor(DFtoplotall$Chromosome)
countspergroup <- data.frame(cases=rowSums(cpm[,dds_filt$group =="CD"])/sum(dds_filt$group =="CD"),
controls=rowSums(cpm[,dds_filt$group =="CTRL"])/sum(dds_filt$group =="CTRL"))
DFtoplotall$MeanCD = log2(countspergroup[,"cases"]+1)
DFtoplotall$MeanCTRL = log2(countspergroup[,"controls"]+1)
DFtoplot = DFtoplotall[DFtoplotall$WaldPvalue_groupCD <= cutoff,]
colnames(DFtoplot) = gsub("WaldPvalue", "-log10_P", colnames(DFtoplot))
targetlist=list(inner=c("-log10_P_contraceptivesyes",
"-log10_P_cigday_1","-log10_P_Age"),
outer=c("MeanCD", "MeanCTRL","-log10_P_groupCD"))
svgfile = paste0(Home,"/output/circos_LME_tags.svg")
topindex=rownames(DFtoplotall)[which(res$pvalue %in% sort(res$pvalue, decreasing = F)[1:10])]
plotcircos(plotdata =DFtoplot, targets = targetlist,
labcol="gene",
title="Differentially Methylated Tag",
pvalident = "log10_P", #collabel for pval identification
pvallog=FALSE, # is log transformed
cutoffpval = 8, #after this cutoff will be black
labelsidx=topindex,
filename = svgfile)
RCircos.Core.Components initialized.
Type ?RCircos.Reset.Plot.Parameters to see how to modify the core components.pdf
2 
#plot best hit
# settings Genomic annotation
scheme <- getScheme()
scheme$GdObject$background.panel = "white"
scheme$GdObject$background.title = "white"
scheme$GdObject$fontcolor.title = "black"
scheme$AnnotationTrack$featureAnnotation = NULL
scheme$AnnotationTrack$fill = "gray"
scheme$AnnotationTrack$fontcolor.item = "black"
scheme$AnnotationTrack$cex = 0.5
scheme$DataTrack$col = Dark8
scheme$GeneRegionTrack$fill <- "dodgerblue3"
scheme$GeneRegionTrack$col <- NULL
scheme$GeneRegionTrack$transcriptAnnotation <- NULL
window=2000
index = which(res$pvalue == min(res$pvalue))
subtitel=paste0(DFtoplotall$Chromosome[index],": ", DFtoplotall$chromStart[index])
chr=DFtoplotall$Chromosome[index]
from = DFtoplotall$chromStart[index]-window
to = DFtoplotall$chromEnd[index]+window
targetrange = GRanges(seqnames = chr,
IRanges(from,
to))
tbl.gene = NULL
attempt = 0
while(is.null(tbl.gene) & attempt<10){
print(paste("tbl.gene attempt:", attempt+1))
if (attempt != 0){
Sys.sleep(60)}
try(tbl.gene <- getTable(ucscTableQuery(mySession,
track="refSeqComposite",
range=targetrange,
table="ncbiRefSeq")))
attempt = attempt+1
}[1] "tbl.gene attempt: 1"tbl.cpg = NULL
attempt = 0
while(is.null(tbl.cpg) & attempt<10){
print(paste("tbl.cpg attempt:", attempt+1))
if (attempt != 0){
Sys.sleep(60)}
try(tbl.cpg <- getTable(ucscTableQuery(mySession,
track="cpgIslandExt",
range=targetrange,
table="cpgIslandExt")))
attempt = attempt+1
}[1] "tbl.cpg attempt: 1"tbl.TFBdg = NULL
attempt = 0
while(is.null(tbl.TFBdg) & attempt<10){
print(paste("tbl.TFBdg attempt:", attempt+1))
if (attempt != 0){
Sys.sleep(60)}
try(tbl.TFBdg <- getTable(ucscTableQuery(mySession,
track="tfbsConsSites",
range=targetrange,
table="tfbsConsSites")))
attempt = attempt+1
}[1] "tbl.TFBdg attempt: 1"tbl.gene_GR <- convertUCSCtoGR(tbl.gene, col.start = "txStart",col.end = "txEnd", col.strand = "strand")
tbl.cpg_GR <- convertUCSCtoGR(tbl.cpg)
tbl.TFBdg_GR <- convertUCSCtoGR(tbl.TFBdg)
itrack <- IdeogramTrack(genome = "hg19", chromosome = as.character(DFtoplotall$Chromosome[index]))
atrack <- GenomeAxisTrack()
dataplot=subsetByOverlaps(GRresultslme_table, targetrange)
indexreads=findOverlaps(GRresultslme_table, targetrange)
if(length(indexreads)>1){
targetcpm=log2_cpm[indexreads@from,]
tmp = dataplot
values(tmp)=targetcpm} else {
targetcpm=t(as.dataframe(log2_cpm[indexreads@from,]))
tmp = dataplot
values(tmp)=targetcpm
}
addScheme(scheme, "myScheme")
options(Gviz.scheme = "myScheme")
ptrack <- DataTrack(dataplot, data = -log10(dataplot$WaldPvalue_groupCD), baseline=0,
name = "P_group", type=c("histogram"), fill="black", col = "black",
col.baseline = "grey")
dtrack <- DataTrack(tmp, groups=Patdata$group,
name = "mean log2(cpm) [SD])", c("heatmap"))
displayPars(dtrack) <- list(type=c("a","confint"))
tbl.gene_GR$symbol = tbl.gene_GR$name2
genotrack <- GeneRegionTrack(tbl.gene_GR, name = "genes", transcriptAnnotation="symbol")
tbl.cpg_GR$symbol = tbl.cpg_GR$name
cpgtrack <- GeneRegionTrack(tbl.cpg_GR, name = "GpG", transcriptAnnotation = "symbol")
displayPars(cpgtrack)<- list(col="white", fill =Dark8[5])
values(tbl.TFBdg_GR)$symbol = tbl.TFBdg_GR$name
tfbtrack <- GeneRegionTrack(tbl.TFBdg_GR, name = "TF-Sites", transcriptAnnotation = "symbol")
displayPars(tfbtrack)<- list(col="white", fill =Dark8[6])
ncols <- 2
nrows <- 1
grid.newpage()
pushViewport(viewport(layout = grid.layout(nrows, ncols, widths=c(1,2))))
pushViewport(viewport(layout.pos.col = 2,layout.pos.row = 1))
p2 = plotTracks(list(itrack,atrack, genotrack, dtrack, ptrack, cpgtrack, tfbtrack), from = from, to = to, sizes=c(1,1,1,4,2,1,4), add=TRUE)
upViewport()
pushViewport(viewport(layout.pos.col = 1,layout.pos.row = 1))
tmp = data.frame(log2_cpm = log2_cpm[index, ], group=Patdata$group)
p1 = ggplot(tmp, aes(x=group, y=log2_cpm, fill=group)) + geom_boxplot(show.legend=T, aes(fill=group)) +
geom_point(position=position_jitterdodge(jitter.width=0.5, dodge.width = 0.3))+scale_fill_manual(values = c("CTRL" = Dark8[1],"CD"=Dark8[2]))
grid.draw(as.grob(p1))
upViewport()
grid.export(paste0(Home,"/output/LME_tophit.svg"))Warning in grabDL(warn, wrap, wrap.grobs, ...): one or more grobs overwritten
(grab WILL not be faithful; try 'wrap.grobs = TRUE')Warning in svgStyleAttributes(style, svgdev): Removing non-SVG style attribute
name(s): subscripts, group.number, group.value, span, degree, family, evaluation
Warning in svgStyleAttributes(style, svgdev): Removing non-SVG style attribute
name(s): subscripts, group.number, group.value, span, degree, family, evaluation

Plot DMR results
DFtoplotall = resultsdmr_table
colnames(DFtoplotall)[1:3] = c("Chromosome", "chromStart", "chromEnd")
DFtoplotall = DFtoplotall[order(DFtoplotall$Chromosome, DFtoplotall$chromStart),]
DFtoplotall$Chromosome = as.factor(DFtoplotall$Chromosome)
DFtoplot = DFtoplotall[(DFtoplotall$p.value<=cutoff | DFtoplotall$p.valueArea<cutoff), ]
targetlist=list(inner=c("p.value",
"p.valueArea"),
outer=c("clusterL"))
svgfile = paste0(Home,"/output/circos_DMR_tags.svg")
topindex=rownames(DFtoplot)[which(DFtoplot$p.value %in% sort(DFtoplot$p.value, decreasing = F)[1:10])]
plotcircos(plotdata =DFtoplot, targets = targetlist,
labcol="name",
title="Differentially Methylated Regions",
pvalident = "p.value", #collabel for pval identification
pvallog=FALSE, # is log transfromed
cutoffpval = 8, #after this cutoff will be black
labelsidx=topindex,
filename = svgfile)
RCircos.Core.Components initialized.
Type ?RCircos.Reset.Plot.Parameters to see how to modify the core components.pdf
2 window=2000
index = which.min(resultsdmr_table$p.value)
chr=resultsdmr_table$chr[index]
from = resultsdmr_table$start[index]-window
to = resultsdmr_table$end[index]+window
targetrange = GRanges(seqnames = chr,
IRanges(from,
to))
tbl.gene <- getTable(ucscTableQuery(mySession, track="refSeqComposite",range=targetrange, table="ncbiRefSeq"))
tbl.cpg <- getTable(ucscTableQuery(mySession, track="cpgIslandExt",range=targetrange, table="cpgIslandExt"))
tbl.TFBdg <- getTable(ucscTableQuery(mySession, track="tfbsConsSites",range=targetrange, table="tfbsConsSites"))
tbl.gene_GR <- convertUCSCtoGR(tbl.gene, col.start = "txStart",col.end = "txEnd", col.strand = "strand")
tbl.cpg_GR <- convertUCSCtoGR(tbl.cpg)
tbl.TFBdg_GR <- convertUCSCtoGR(tbl.TFBdg)
itrack <- IdeogramTrack(genome = "hg19", chromosome = as.character(chr))
atrack <- GenomeAxisTrack()
dataplot=subsetByOverlaps(GRresultslme_table, targetrange)
indexreads=findOverlaps(GRresultslme_table, targetrange)
selected = which.min(as.data.frame(GRresultslme_table)[indexreads@from, "WaldPvalue_groupCD"])
if(length(indexreads)>1){
targetcpm=log2_cpm[indexreads@from,]
tmp = dataplot
values(tmp)=targetcpm} else {
targetcpm=t(as.dataframe(log2_cpm[indexreads@from,]))
tmp = dataplot
values(tmp)=targetcpm
}
ptrack <- DataTrack(dataplot, data = -log10(dataplot$WaldPvalue_groupCD), baseline=0,
name = "P_group", type=c("histogram"), fill="black", col = "black",
col.baseline = "grey")
dtrack <- DataTrack(tmp, groups=Patdata$group,
name = "mean log2(cpm) [SD])", c("heatmap"))
displayPars(dtrack) <- list(type=c("a","confint"))
tbl.gene_GR$symbol = tbl.gene_GR$name2
genotrack <- GeneRegionTrack(tbl.gene_GR, name = "genes", transcriptAnnotation="symbol")
tbl.cpg_GR$symbol = tbl.cpg_GR$name
cpgtrack <- GeneRegionTrack(tbl.cpg_GR, name = "GpG", transcriptAnnotation = "symbol")
displayPars(cpgtrack)<- list(col="white", fill =Dark8[5])
values(tbl.TFBdg_GR)$symbol = tbl.TFBdg_GR$name
tfbtrack <- GeneRegionTrack(tbl.TFBdg_GR, name = "TF-Sites", transcriptAnnotation = "symbol")
displayPars(tfbtrack)<- list(col="white", fill =Dark8[6])
ncols <- 2
nrows <- 1
grid.newpage()
pushViewport(viewport(layout = grid.layout(nrows, ncols, widths=c(1,2))))
pushViewport(viewport(layout.pos.col = 2,layout.pos.row = 1))
p2 = plotTracks(list(itrack,atrack, genotrack, dtrack, ptrack, cpgtrack, tfbtrack),
from = from, to = to, sizes=c(1,2,2,4,2,1,4), add=T)
upViewport()
pushViewport(viewport(layout.pos.col = 1,layout.pos.row = 1))
tmp = data.frame(log2_cpm = log2_cpm[indexreads@from[selected],], group=Patdata$group)
p1 = ggplot(tmp, aes(x=group, y=log2_cpm, fill=group)) + geom_boxplot(show.legend=T, aes(fill=group)) +
geom_point(position=position_jitterdodge(jitter.width=0.5, dodge.width = 0.3))+scale_fill_manual(values = c("CTRL" = Dark8[1],"CD"=Dark8[2]))
grid.draw(as.grob(p1))
upViewport()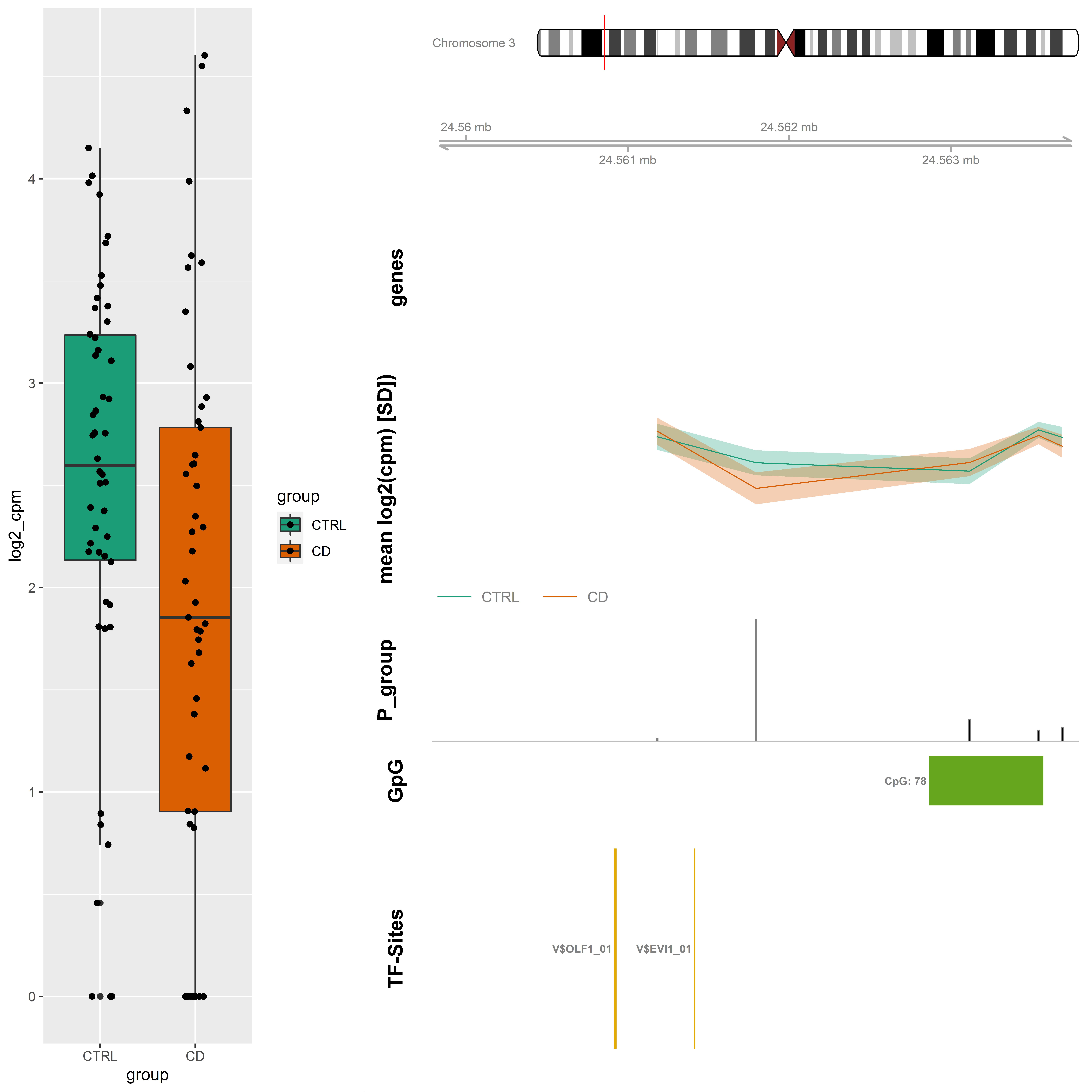

sessionInfo()R version 4.0.3 (2020-10-10)
Platform: x86_64-w64-mingw32/x64 (64-bit)
Running under: Windows 10 x64 (build 19042)
Matrix products: default
locale:
[1] LC_COLLATE=German_Germany.1252 LC_CTYPE=German_Germany.1252
[3] LC_MONETARY=German_Germany.1252 LC_NUMERIC=C
[5] LC_TIME=German_Germany.1252
attached base packages:
[1] grid parallel stats4 stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] scales_1.1.1 compareGroups_4.4.6
[3] forcats_0.5.0 stringr_1.4.0
[5] dplyr_1.0.2 purrr_0.3.4
[7] readr_1.4.0 tidyr_1.1.2
[9] tibble_3.0.4 ggplot2_3.3.3
[11] tidyverse_1.3.0 kableExtra_1.3.1
[13] gridSVG_1.7-2 ggplotify_0.0.5
[15] rtracklayer_1.49.5 Gviz_1.34.0
[17] RColorBrewer_1.1-2 beeswarm_0.2.3
[19] RCircos_1.2.1 DESeq2_1.30.0
[21] SummarizedExperiment_1.20.0 Biobase_2.50.0
[23] MatrixGenerics_1.2.0 matrixStats_0.57.0
[25] GenomicRanges_1.42.0 GenomeInfoDb_1.26.2
[27] IRanges_2.24.1 S4Vectors_0.28.1
[29] BiocGenerics_0.36.0 workflowr_1.6.2
loaded via a namespace (and not attached):
[1] uuid_0.1-4 readxl_1.3.1 backports_1.2.0
[4] Hmisc_4.4-2 systemfonts_0.3.2 BiocFileCache_1.14.0
[7] lazyeval_0.2.2 splines_4.0.3 BiocParallel_1.24.1
[10] digest_0.6.27 ensembldb_2.14.0 htmltools_0.5.1.1
[13] fansi_0.4.1 Rsolnp_1.16 magrittr_2.0.1
[16] checkmate_2.0.0 memoise_2.0.0 BSgenome_1.58.0
[19] cluster_2.1.0 Biostrings_2.58.0 annotate_1.68.0
[22] modelr_0.1.8 officer_0.3.16 askpass_1.1
[25] prettyunits_1.1.1 jpeg_0.1-8.1 colorspace_2.0-0
[28] blob_1.2.1 rvest_0.3.6 rappdirs_0.3.1
[31] haven_2.3.1 xfun_0.20 crayon_1.3.4
[34] RCurl_1.98-1.2 jsonlite_1.7.2 genefilter_1.72.0
[37] survival_3.2-7 VariantAnnotation_1.36.0 glue_1.4.2
[40] gtable_0.3.0 zlibbioc_1.36.0 XVector_0.30.0
[43] webshot_0.5.2 DelayedArray_0.16.0 DBI_1.1.1
[46] Rcpp_1.0.5 viridisLite_0.3.0 xtable_1.8-4
[49] progress_1.2.2 htmlTable_2.1.0 gridGraphics_0.5-1
[52] foreign_0.8-81 bit_4.0.4 Formula_1.2-4
[55] truncnorm_1.0-8 htmlwidgets_1.5.3 httr_1.4.2
[58] ellipsis_0.3.1 mice_3.12.0 farver_2.0.3
[61] pkgconfig_2.0.3 XML_3.99-0.5 nnet_7.3-15
[64] dbplyr_2.0.0 locfit_1.5-9.4 labeling_0.4.2
[67] tidyselect_1.1.0 rlang_0.4.10 later_1.1.0.1
[70] AnnotationDbi_1.52.0 cellranger_1.1.0 munsell_0.5.0
[73] tools_4.0.3 cachem_1.0.1 cli_2.2.0
[76] generics_0.1.0 RSQLite_2.2.2 broom_0.7.3
[79] evaluate_0.14 fastmap_1.1.0 yaml_2.2.1
[82] knitr_1.30 bit64_4.0.5 fs_1.5.0
[85] zip_2.1.1 AnnotationFilter_1.14.0 whisker_0.4
[88] xml2_1.3.2 biomaRt_2.46.2 compiler_4.0.3
[91] rstudioapi_0.13 curl_4.3 png_0.1-7
[94] reprex_1.0.0 geneplotter_1.68.0 HardyWeinberg_1.7.1
[97] stringi_1.5.3 ps_1.5.0 GenomicFeatures_1.42.1
[100] gdtools_0.2.3 lattice_0.20-41 ProtGenerics_1.22.0
[103] Matrix_1.2-18 vctrs_0.3.6 pillar_1.4.7
[106] lifecycle_0.2.0 BiocManager_1.30.10 flextable_0.6.2
[109] data.table_1.13.6 bitops_1.0-6 httpuv_1.5.5
[112] R6_2.5.0 latticeExtra_0.6-29 promises_1.1.1
[115] gridExtra_2.3 writexl_1.3.1 dichromat_2.0-0
[118] assertthat_0.2.1 chron_2.3-56 openssl_1.4.3
[121] rprojroot_2.0.2 withr_2.4.1 GenomicAlignments_1.26.0
[124] Rsamtools_2.6.0 GenomeInfoDbData_1.2.4 hms_1.0.0
[127] rpart_4.1-15 rmarkdown_2.6 rvcheck_0.1.8
[130] git2r_0.28.0 biovizBase_1.38.0 lubridate_1.7.9.2
[133] base64enc_0.1-3